
神经肌肉病概述
神经肌肉病(neuromuscular disorder,NMD)是一组影响肌肉功能的疾病统称,可由随意肌的直接病变,也可因神经或神经肌肉接头的间接损伤所致,通常可引起进行性肌无力。神经肌肉病可分类为不同的临床类型,其诊断依赖于临床表现、肌电图(EMG)、肌肉活检病理、生化检测、基因检测等辅助检查。不同的疾病类型可见于不同的年龄阶段,新生儿期到成年期。遗传性神经肌肉病的患病率约为1/3500。
绝大多数神经肌肉病是由基因变异引起的,并在疾病遗传模式、基因变异类型、发病率、临床症状、发病年龄、疾病进展速度、预后等方面存在多样性。神经肌肉接头的问题可导致眼睑下垂、复视、疲乏加剧等。

神经肌肉病诊断策略
常规诊断程序中,神经肌肉病依赖于神经电生理和肌肉活检措施,但这类疾病,也许可以考虑更早的基因检测,有助于减少非必要的临床检测工作[PMID:24588502]。
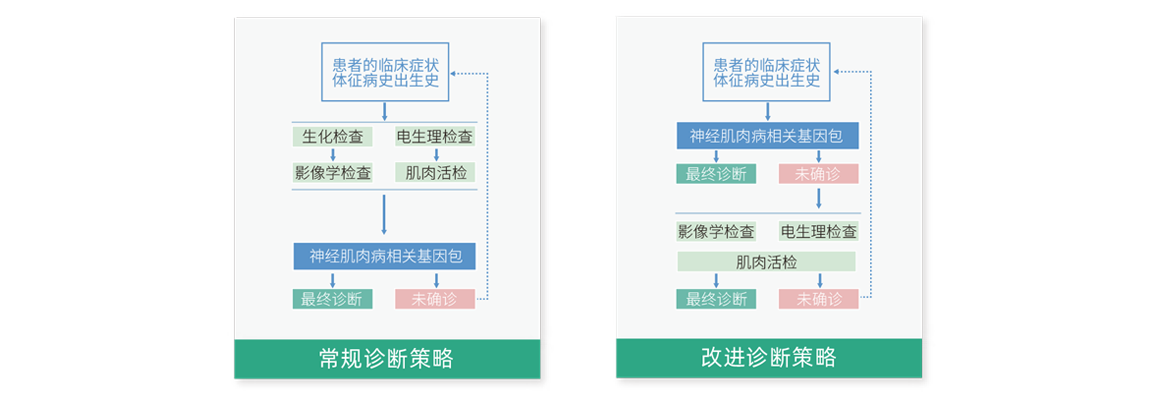
嘉检神经肌肉病检测基因包
嘉检神经肌肉病优选基因包检测包括高肌酸激酶血症、发作性运动诱发性运动障碍、肌营养不良、代谢性肌病、腓骨肌萎缩症等疾病。可同时分析SNV和indel(包括已知致病内含子变异)、拷贝数变异(CNV)、AOH,尤其是新型变异、罕见变异及嵌合变异等。

注:基因包内的所有基因均可以单独检测,并根据患者的临床表型进行调整,且随着医学的发展而有所变化,详情可咨询400-179-2998。
精典病例 - 肢带型肌营养不良
检测结果质控统计:这个综合征基因包检测区间包括2,122个相关基因,30,309个编码区总共含4,943,159个碱基。
平均覆盖深度287+/-146X,大于10X覆盖区间占99.9%,大于20X覆盖区间占99.7%。
| 基因名称 | OMIM编号 | 遗传方式 | HG19位置 | 转录本 | 基因区 | 核苷酸与氨基酸改变 | 合子状态 | 人群频率 | ACMG变异 分类 | 相关疾病/文献 | 来源 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DYSF | 603009 | AR | chr2:7190xxxx | NM_001130987 | - | c.xxxx-1G>A | 杂合 | - | 2类-可能致病 | 肢带型肌营养不良2B型 | 父亲(杂合) |
| DYSF | 603009 | AR | chr2:7178xxxx-7178xxxx | NM_001130987 | - | 23号外显子缺失 | x1 | - | 2类-可能致病 | 肢带型肌营养不良2B型 | 母亲(杂合) |
① 一般成年起病,CK增高,早期表现为四肢肌无力,肩胛肌萎缩等。
精典病例 - 腓骨肌萎缩症
检测结果质控统计:这个综合征基因包检测区间包括2,122个相关基因,30,309个编码区总共含4,943,159个碱基。
平均覆盖深度295+/-156X,大于10X覆盖区间占99.9%,大于20X覆盖区间占99.7%。
| 基因名称 | OMIM编号 | 遗传方式 | HG19位置 | 转录本 | 基因区 | 核苷酸与氨基酸改变 | 合子状态 | 人群频率 | ACMG变异 分类 | 相关疾病/文献 | 来源 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PMP22 | 300377 | AD | chr17:1513xxxx-1516xxxx | NM_000304 | - | 1-4号外显子重复 | - | - | 1类-致病变异 | 腓骨肌萎缩症 | 新发 |
① 四肢远端进行性肌无力,肌萎缩和感觉障碍;
② 通常伴有神经传导速度减慢,高足弓,足下垂等,有明显的遗传异质性。